methylQA is aimed to be a methylation sequencing data quality assessment tool for MeDIP-seq and MRE-seq. It provides basic mapping status of the next generating sequencing data, like number of total reads, number of mapped reads etc., it also provide CpG status like how many CpG have been covered by one experiment, how many times one CpG have been covered etc. methylQA could also process general ChIP-seq data like Histone/TF ChIP-seq data, generate read density and mapping statistics.
methylQA could generate a new and comprehensive view of whole genome bisulfite sequencing data for
WashU EpiGenome Browser,
it's called methylC track,
which contains read density, CpG methylation, CHG methylation and CHH methylation
information on forward/reverse strand. For more about this track type, please go to this link.
An example view is shown below:
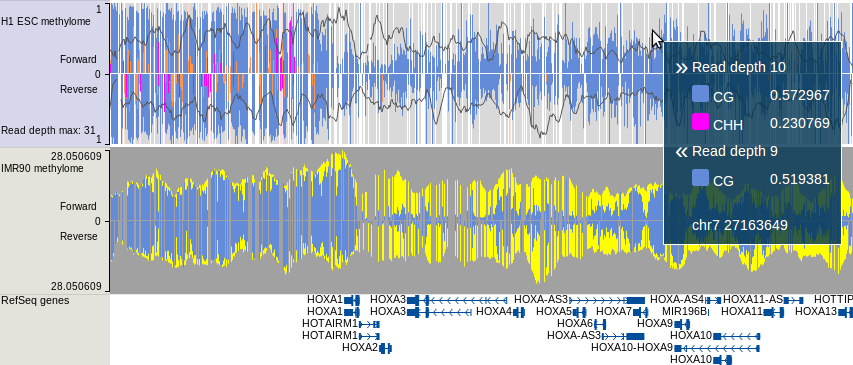 For details of how to generate this track, please see the bismark section of Tutorial.
For details of how to generate this track, please see the bismark section of Tutorial.
methylQA used a lot of functions from UCSC Kent source and samtools, see Install for installation instructions, and see Tutorial for documentations and tutorials.
Questions or comments, please contact our mailing list, thanks!
Citation
If you find methylQA useful, could you please cite:
Li et.al, Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods, 2014