Contents
- Process MeDIP-seq data using
medipsubcommand - Process MRE-seq data using
mresubcommand - Process ChIP-seq data using
densitysubcommand - Process Bismark generated Whole Genome Bisulfite sequencing data using
bismarksubcommand
Subcommands medip, mre and density accept BAM/SAM files generated by BWA as input,
and bismark accept BAM/SAM files generated by Bismark as input.
Process MeDIP-seq data using medip subcommand [Top]
The medip subcommand is used to generate genome density for MeDIP-Seq data,
and reads mapping and CpG coverage statistics. An example command would look like:
methylQA medip -m /path/of/CpG_no_contig.bed /path/of/hg19_lite.size sample_medip.bam
Note, the CpG bed file (-m parameter) is optional, if not specified, will not generate CpG statistics.
This would generate some files, the most useful file would be:
sample_medip.bigWig (read density, could be upload to Genome Browser) sample_medip.extended.bed (an alignment in bed format) sample_medip.pdf (a PDF report of this data) sample_medip.report (a TEXT report of this data)
The PDF report file contains statistics of reads mapping, CpG coverage etc. Example figures are illustrated below:
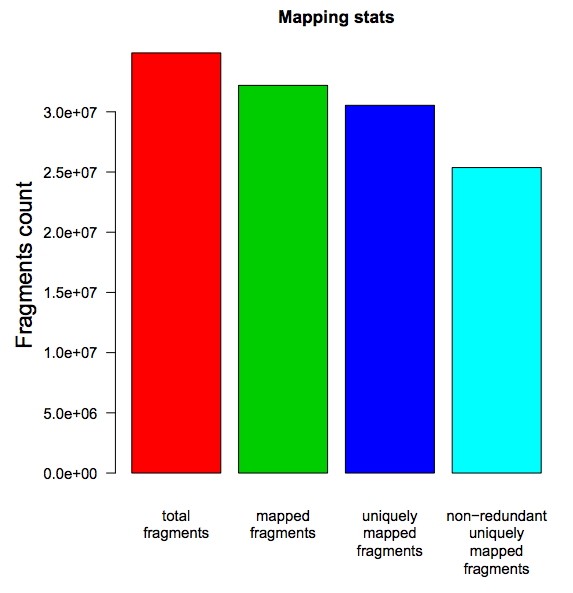
Reads mapping stats:
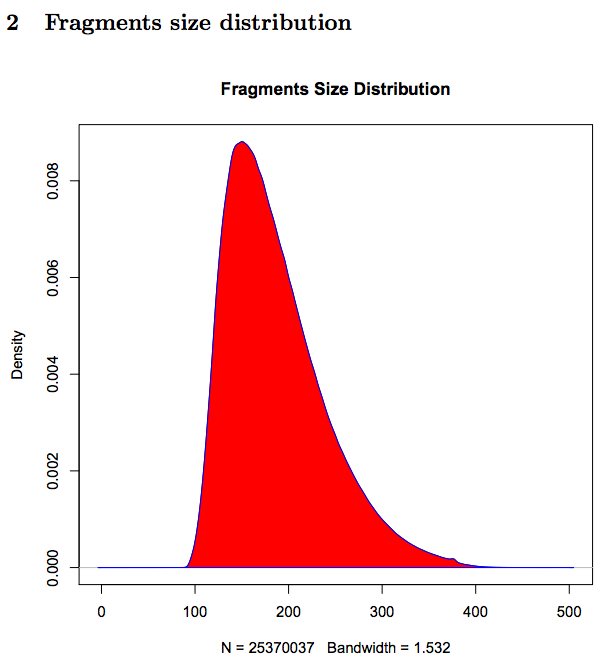
Fragments size distribution (only for Paired-end sequencing data):
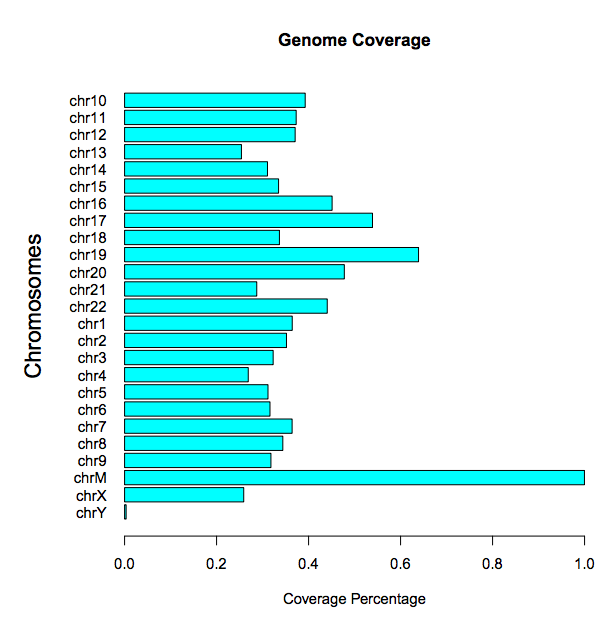
Genomic coverage:
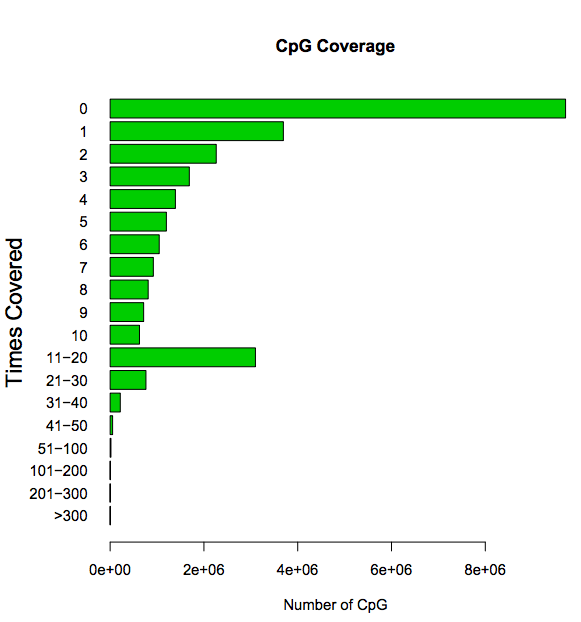
CpG coverage:
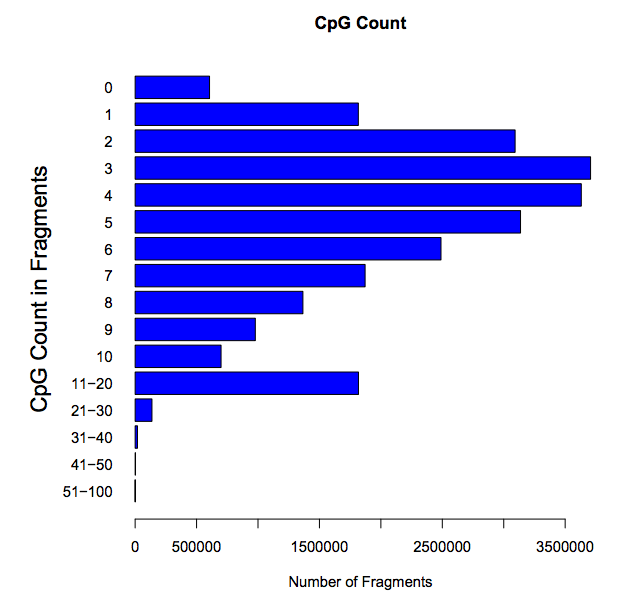
CpG count:
Process MRE-seq data using mre subcommand [Top]
The mre subcommand is used to generate CpG density for MRE-Seq data,
and reads mapping, MRE cutting and CpG coverage statistics. An example command would look like:
methylQA medip -m /path/of/CpG_no_contig.bed /path/of/hg19_lite.size /path/of/FiveMRE_frags.bed sample_mre.bam
Note, the CpG bed file (-m parameter) is optional, if not specified, will not generate CpG statistics.
This would generate some files, the most useful file would be:
sample_mre.CpG.bigWig (CpG density, could be upload to Genome Browser) sample_mre.filer.bed (filtered alignment with MRE sites in bed format) sample_mre.pdf (a PDF report of this data) sample_mre.CpG.report (a TEXT report of this data)
The PDF report file contains statistics of reads mapping, CpG coverage etc. Example figures are illustrated below:
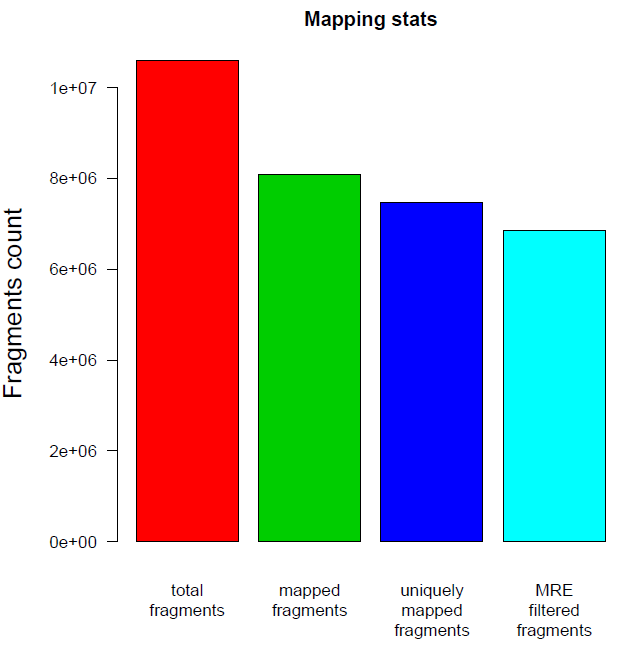
Reads mapping stats:
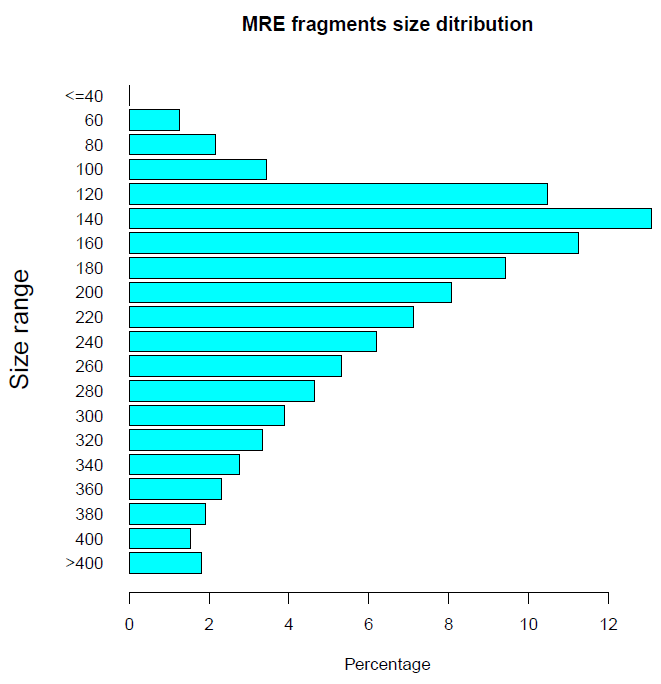
Distribution by enzyme cut site:
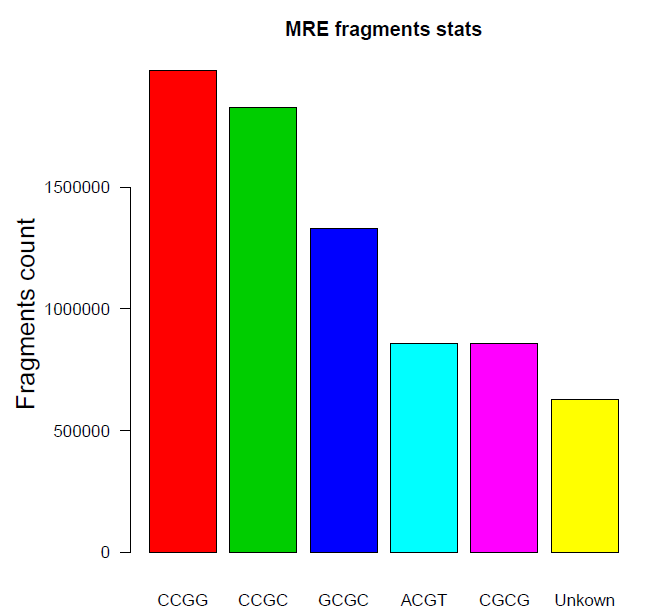
MRE fragments stats:
Process ChIP-seq data using density subcommand [Top]
For other ChIP-Seq based data, The density subcommand is used to generate reads density,
and reads mapping statistics. An example command would look like:
methylQA density /path/of/hg19_lite.size sample_chipseq.bam
This would generate some files, the most useful file would be:
sample_density.bigWig (read density, could be upload to Genome Browser) sample_chipseq.extended.bed (an alignment in bed format) sample_chipseq.report (a TEXT report of this data)
Process Bismark generated Whole Genome Bisulfite sequencing data using bismark subcommand [Top]
For processing whole genome bisulfite sequencing data,
the raw reads need to be aligned by Bismark first (v0.9.0 recommended),
then the bismark subcommand is used to analyze Bismark output.
This module has been tested with version v0.7.6 and v0.9.0 of Bismark. Support of other bisulfite callers (BS-Seeker, BSMap, Methylcoder, etc.) are under developing.
Note, currently this subcommand works only for Bismark + Bowtie1.
This subcommand have 3 running modes:
CpG mode (default)
The default mode of this subcommand would generated statistics of each CpG site. An example command would look like:
methylQA bismark /path/of/hg19_lite.size /path/of/CpG_no_contig.bed sample_bismark_1.bam,sample_bismark_2.bam
This would generate some files, the most useful file would be:
sample_bismark1_CpG.bedGraph (CpG methylation percentage in bedGraph format) sample_bismark1_density.bedGraph (CpG reads density in bedGraph format) sample_bismark1.report (a TEXT report of this data)
Note: when use multiple BAM/SAM files as input, the files need separated by comma, and the default output prefix would be the 1st input filename without suffix. (could use -o parameter to overwrite)
The bedGraph files could be either processed by tabix for WashU EpiGenome Browser, or be converted to bigWig for UCSC Genome Browser.
An example .report file would look like:
files provided: 4
sample_1.bam
sample_2.bam
sample_3.bam
sample_4.bam
uniquely mappable reads (pair): 814393162
quality failed mapped reads (pair) in the bismark bam: 52124490
total base of uniquely mapped reads (pair): 81439316200
total base of uniquely mapped reads (pair) cover genome base (3095677412): 26.3X
in all uniquely mapped reads (pair), found:
number of methylated C in CHG context (was protected): 17749762
number of not methylated C in CHG context (was converted): 2236057612
C->T convertion rate in CHG context: 99.21%
number of methylated C in CHH context (was protected): 60092628
number of not methylated C in CHH context (was converted): 7841475114
C->T convertion rate in CHH context: 99.24%
number of methylated C in CpG context (was protected): 298994806
number of not methylated C in CpG context (was converted): 160192606
C->T convertion rate in CpG context: 34.89%
number of methylated C in Unknown context (was protected): 0
number of not methylated C in Unknown context (was converted): 0
C->T convertion rate in Unknown context: -nan%
in the total 28217009 CpG:
Times covered Count Percent |
0 513313 1.82 |*
1 305342 1.08 |*
2 369204 1.31 |*
3 479072 1.70 |*
4 597323 2.12 |**
5 721190 2.56 |**
6 839913 2.98 |**
7 954587 3.38 |***
8 1063685 3.77 |***
9 1157370 4.10 |****
10 1233325 4.37 |****
11-20 12213251 43.28 |*******************************************
21-30 5972629 21.17 |*********************
31-40 1577836 5.59 |*****
41-50 185637 0.66 |
51-100 24314 0.09 |
101-200 3801 0.01 |
201-300 1252 0.00 |
>300 3965 0.01 |
CpG Cytosine mode (-b parameter)
This subcommand could generated statistics of each Cytosine of CpG sites by specify -b option. An example command would look like:
methylQA bismark -b /path/of/hg19_lite.size /path/of/CpG_no_contig.bed sample_bismark_1.bam,sample_bismark_2.bam
Note, the output and report files would be similar as above, but based on single C base.
Full mode (-F parameter)
The full mode of this subcommand would generated statistics of each Cytosine (CpG, CHG, CHH). An example command would look like:
methylQA bismark -F /path/of/hg19_lite.size /path/of/CpG_no_contig.bed sample_bismark_1.bam,sample_bismark_2.bam
Note, the full mode need quite a lot of memory and long running time.
We would show an example of this full mode using the IMR90 WGBS data from Roadmap Epigenomics Project, the IMR90 datasets contains 2 replicates, and each replicate contain 3 parts of data. Download the raw data from NCBI SRA database, align them to Human hg19 using lastest version of Bismark. After alignment, we could get 6 Bismark aligned BAM/SAM files, then we run following command:
methylQA bismark -F /path/of/hg19_lite.size /path/of/CpG_no_contig.bed SRX006786.fq_bismark.bam,SRX006788.fq_bismark.bam,SRX006785.fq_bismark.bam,SRX006787.fq_bismark.bam,SRX006784.fq_bismark.bam,SRX006783.fq_bismark.bam -o IMR90
Note, this step need about 110G memory and takes about 12 hours.
This would generate 8 track files and 1 report file:
IMR90.report (a TEXT report of this data) IMR90.reverse.Density.bedGraph (read density of reverse strand in bedGraph format) IMR90.forward.Density.bedGraph (read density of forward strand in bedGraph format) IMR90.reverse.CHH.bedGraph (CHH methylation percentage of reverse strand in bedGraph format) IMR90.forward.CHH.bedGraph (CHH methylation percentage of forward strand in bedGraph format) IMR90.reverse.CHG.bedGraph (CHG methylation percentage of reverse strand in bedGraph format) IMR90.forward.CHG.bedGraph (CHG methylation percentage of forward strand in bedGraph format) IMR90.reverse.CG.bedGraph (CpG methylation percentage of reverse strand in bedGraph format) IMR90.forward.CG.bedGraph (CpG methylation percentage of forward strand in bedGraph format)
These 8 bedGraph files could submited to WashU EpiGenome Browser for visualization. For more about this, please go to this link. An example view is shown blow:
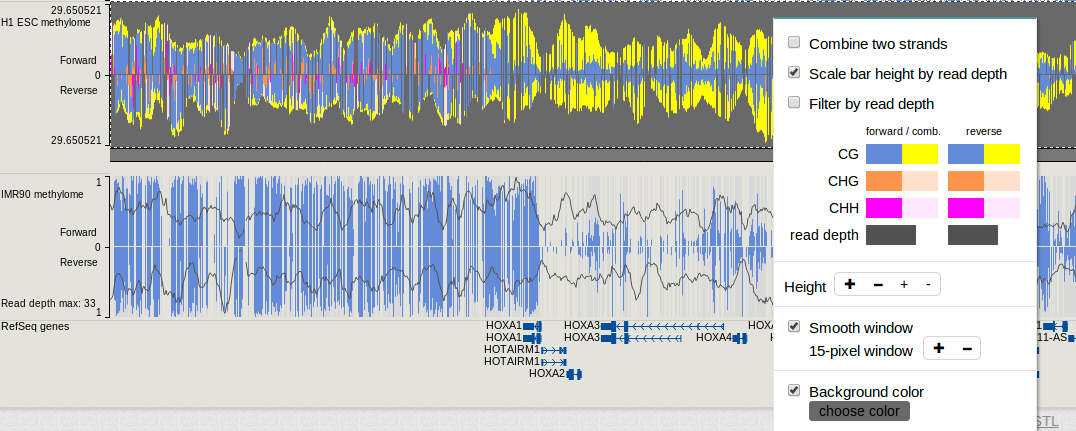
Instructions of how to visualize this methyC track on WashU EpiGenome Browser
- Use bgzip/tabix to compress and index these 8 bedGraph files above, see detailed steps here
- Put all .gz and .gz.tbi file at an Http URL
- Build a text file, contains following contents:
[ { type:"methylC", name:"IMR90 methylome", custom_annotation:['IMR90', 'WGBS'], mode:"show", height:70, combinestrands:false, scalebarheight:false, smoothwindow:11, tracks:{ forward:{ ReadDepth:{color:'#525252',url:'http://your.url.here/IMR90.forward.readDepth.bedGraph.gz'}, CG:{color:'rgb(100,139,216)',bg:'#d9d9d9',url:'http://your.url.here/IMR90.forward.CGmethy.bedGraph.gz'}, CHG:{color:'rgb(255,148,77)',bg:'#ffe0cc',url:'http://your.url.here/IMR90.forward.CHGmethy.bedGraph.gz'}, CHH:{color:'rgb(255,0,255)',bg:'#ffe5ff',url:'http://your.url.here/IMR90.forward.CHHmethy.bedGraph.gz'}, }, reverse:{ ReadDepth:{color:'#525252',url:'http://your.url.here/IMR90.reverse.readDepth.bedGraph.gz'}, CG:{color:'rgb(100,139,216)',bg:'#d9d9d9',url:'http://your.url.here/IMR90.reverse.CGmethy.bedGraph.gz'}, CHG:{color:'rgb(255,148,77)',bg:'#ffe0cc',url:'http://your.url.here/IMR90.reverse.CHGmethy.bedGraph.gz'}, CHH:{color:'rgb(255,0,255)',bg:'#ffe5ff',url:'http://your.url.here/IMR90.reverse.CHHmethy.bedGraph.gz'}, }, } }, {type:'metadata',vocabulary:{ 'Sample':['IMR90'], 'Assay':['WGBS'], }, show:['Sample','Assay'], }, ]put this text file to an HTTP url, for example, http://your.url.here/hub.json - Go to WashU EpiGenome Browser, click 'CustomTK' button, then select 'Datahub'
- Submit this URL: http://your.url.here/hub.json, click 'Submit', done!